|

2011, Vol. 6 No. 1, Article 77
Molecular Characterization and Diversity of Rumen Bacterial Flora in Indian Goat by 16S rDNA Sequencing
Jayesh Kumar Manilal Patel1, Mayur. K. Jhala2,
Prashant Soni1, Nadeem Shabir*1, P. R. Pandya4,
K. M. Singh1, D. N. Rank3 and C. G. Joshi1
1Department
of Animal Biotechnology
2Department of Veterinary Microbiology
3Department of Animal Genetics and Breeding
4Animal Nutrition Research Station
Anand Agricultural University, Anand (388 001), Gujarat, India
*Corresponding Author;
e-mail address: [email protected]
ABSTRACT
Present study was aimed to characterize bacterial communities in goat rumen using a culture-independent approach for a pooled sample of rumen fluid from five adult surti goats. Due to their unique browsing habits including feeding on unconventional tree leaves, goats are expected to host distinct bacterial communities in their rumen. So, 16S rDNA sequences were amplified and cloned from the sample and 60 such clones were sequenced to evaluate the bacterial diversity in goat rumen. Sequence similarity search with Genbank databases revealed that five clones (8%) exhibited similarities with known bacterial species (viz. Ruminococcus albus, Ruminococcus flavefaciens, Prevotella multiformis {2 clones} and Butyrivibrio fibrisolvens) and five clones (8%) showed similarities with known bacterial genera (viz., Ruminococcus, Prevotella, and Bacillus {3 clones}). Taxonomic classification by Ribosomal Database Project (RDP) revealed that the 60 clones mainly belonged to two phyla viz. Bacteriodes (21 clones) and Firmicutes (20 clones), however, 17 clones fell under unclassified bacteria and two clones under unclassified root. Phylogenetic analysis using neighbour-joining method grouped three clones (5%) with species, two clones (3%) with genera, one clone (1.6%) with family, twenty seven clones (45%) with unidentified rumen bacteria and the remaining twenty seven clones (45%) grouped separately and showed distinct genetic grouping compared to the reported sequences in the Genbank database.
KEY
WORDS
Goat rumen, 16S rDNA, clones, sequencing and bacterial diversity.
INTRODUCTION
The ruminants are fed on lignocellulosic agricultural by-products (like cereal straw, tree foliage, etc.) and cakes of oil seeds in tropical countries like India. The digestion of such plant materials occurs by virtue of the extensive microbial community, including bacteria, fungi, and protozoa [1], which are found in the rumen and provide the host with nutrients, predominantly in the form of volatile fatty acids and microbial protein. The microbes present in the rumen ecosystem can be classified into 3 domains: Bacteria, Archaea (methanogens), and Eucarya (protozoa and fungi) [2]. Classically, most of the knowledge of the microbial composition of rumen fluid has been derived using traditional methods, such as the roll-tube technique [3] or most probable number (MPN) estimates [4]. The rumen bacteria have been shown by traditional procedures to belong to some 22 predominant species [5]. Rumen microbes have been extensively studied both qualitatively [6] and quantitatively [7] using DNA-based technologies(16s RNA/ 18s RNA gene). These techniques have been further used to construct a library of 16S rDNA clones of rumen microbes and to demonstrate considerable diversity of rumen bacteria. Molecular research on microbial ecology of animals provides a broad perspective of application in the field of animal sciences [8]. India ranks second in the goat population of world contributing 25.6% to the total livestock population of the country [9]. Since Indian goats primly sustain on grazing and crop residues, the microbial population is expected to be more diverse. Thus the identification of the rumen flora, bacteria in particular, seems important in understanding the digestion potential of unconventional feed in goats. Surti is a popular breed of goat found in central and southern Gujarat. Surti goats are of medium size predominantly white in color, with a body weight of 32-35 kg. The present study includes the identification of rumen bacteria in surti goat by amplification, cloning and sequencing of 16S rRNA gene of bacteria, followed by sequence comparison and phylogenetic analysis.
MATERIALS AND METHODS
Sample collection and DNA extraction
The experiment was carried out on five adult surti goats, at approximately 3 years of age and with a mean live weight of 32±3.2 kg. They were reared at Instructional Farm, College of Veterinary Science and Animal Husbandry, Anand Agricultural University, Anand. All the animals were maintained under a uniform feeding regime [10] for at least 21 days. The diet comprised of green fodder Napier bajra 21 (Pennisetum purpureum), mature pasture grass (Dicanthium annulatum), non-conventional feed (Azadirachta indica) and concentrate mixture (20% crude protein, 65% total digestible nutrients). Approximately 50 ml of rumen fluid from each goat was collected via a stomach tube located in the middle part of the rumen and connected to a vacuum pump at 0, 2, 4, 6 h after feeding [11]. Samples were pooled and filtered through four layers of cheesecloth to remove particulate matter. Strained samples were used for the total microbial DNA extraction. Total DNA (0, 2, 4 and 6 hrs x 5 animals) extraction was done using Phenol: Chloroform method.
PCR amplification of the 16S rRNA gene
Bacteria specific primers [12]: a forward primer 27F (5’- AGAGTTTGATCMTGGCTCAG - 3’) and a reverse primer 1492R (5’- ATAGGYTACCTTGTTACGACT- 3’) were used. Subsquently, 16S rDNA was amplified using the metagenomic DNA and Master mix (Fermentas, UK). A total of 25 µL of reaction mixture consisted of 10 pmol of each primer, 30 ng of template DNA, and 12.5 µL of Master mix (Fermentas, UK). The PCR amplification was performed by Thermal Cycler (ABI, USA) using the following program: denaturing at 95°C for 5 min, followed by 30 cycles of 30 s of denaturing at 95°C, 30 s of annealing at 50°C and 2 min of elongation at 72°C, with a final extension at 72°C for 10 min for the first set. The anticipated product of approximately 1.4 kb was separated by agarose gel electrophoresis and cleaned by using a QIAquick DNA Gel Extraction Kit (QIAGEN, CA) in accordance with the directions of the manufacturer.
Cloning and sequencing
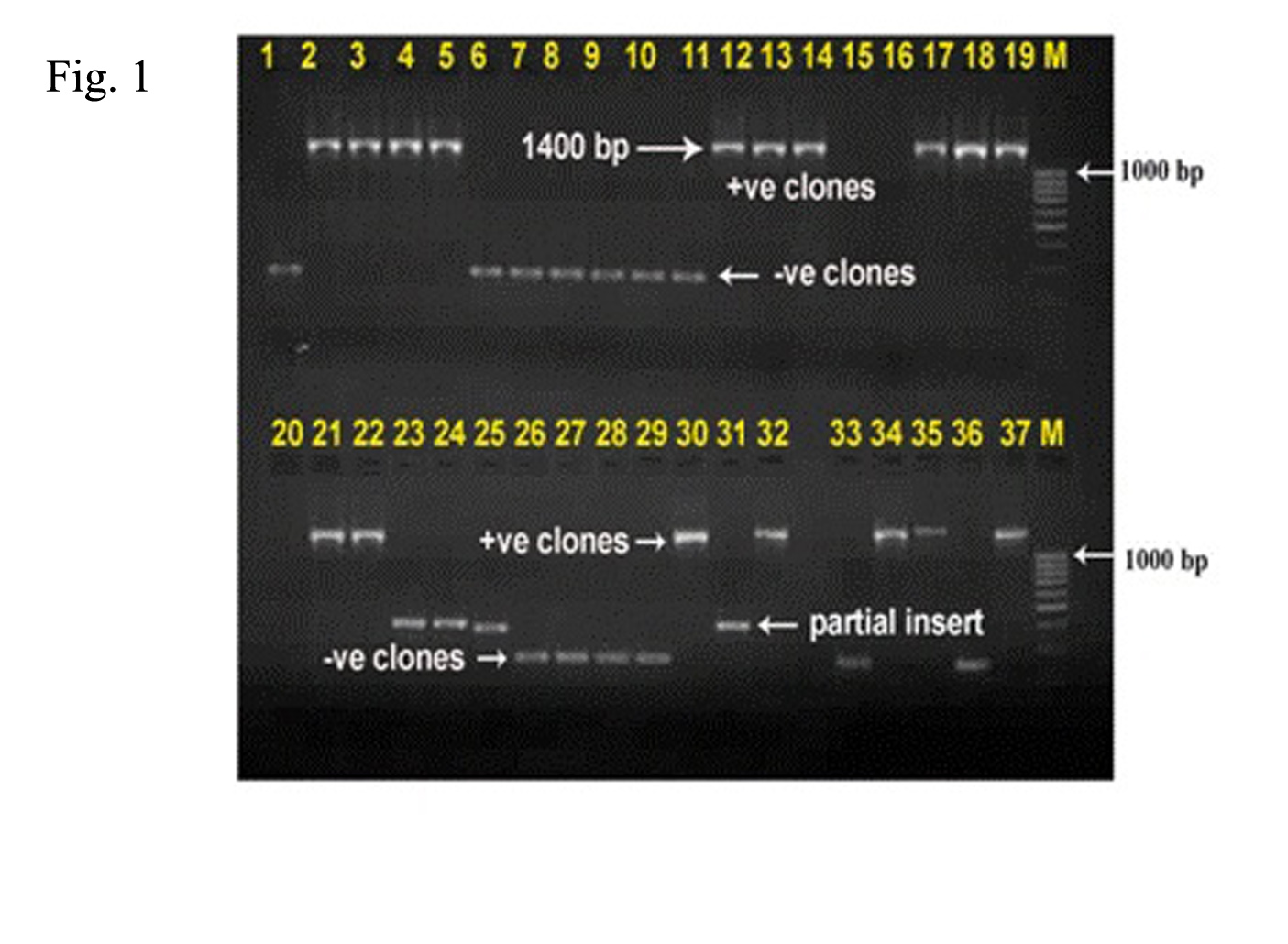
The purified PCR products were cloned in InstaTA cloning kit (Fermentas, UK) according to the instructions of the manufacturer. The recombinant plasmids were then extracted by the Mini-prep Plasmid Extraction Kit (QIAGEN, CA) (Fig. 1). Sequencing was performed for all the clones in the library with an ABI Prism 310 Genetic Analyser (Applied Biosystems Inc., CA) using BigDye Terminator (version 3.1) in the Animal Biotechnology Laboratory, AAU, Anand, Gujarat, India.
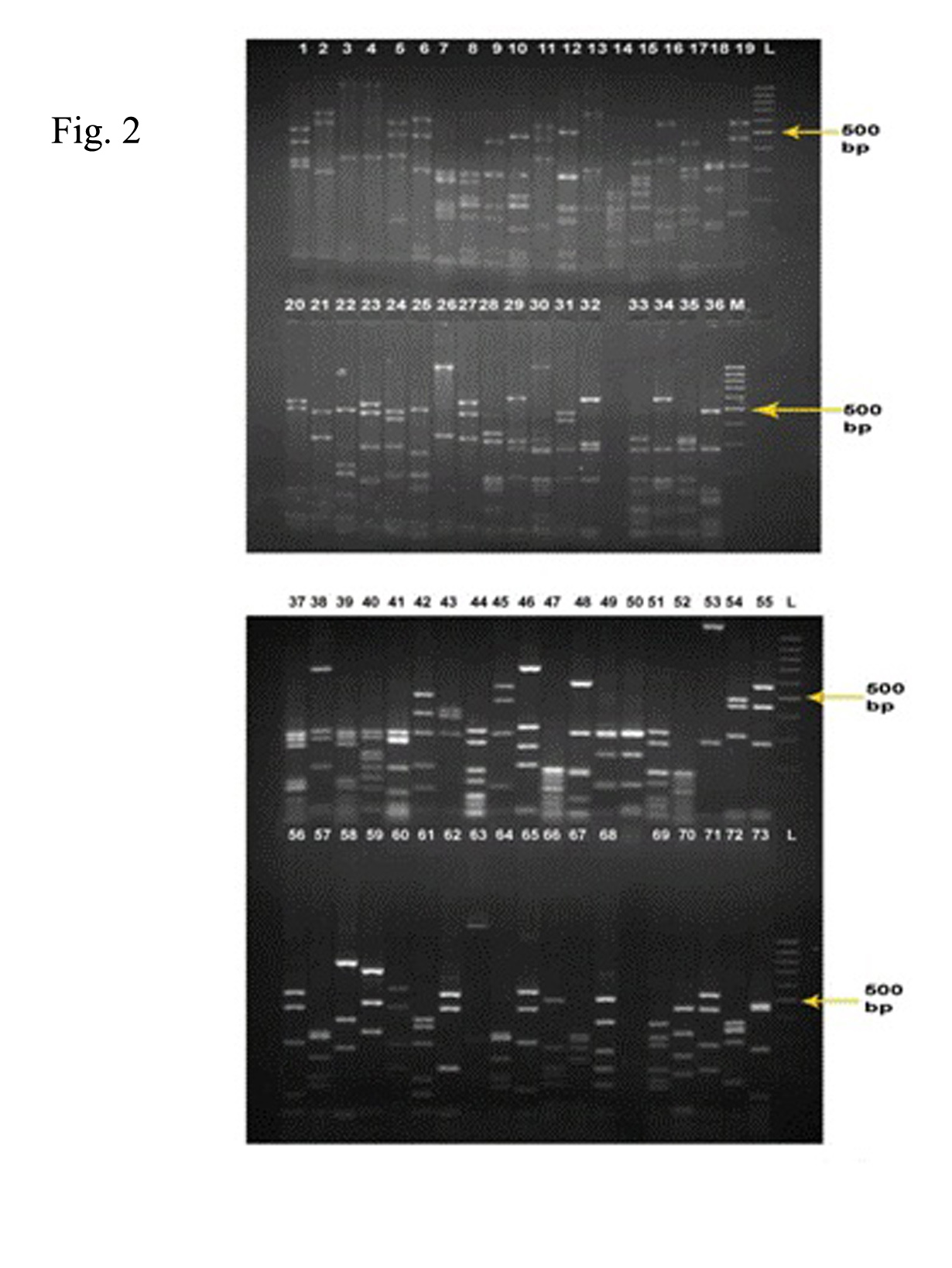
Restriction digestion of the colony PCR products (73 clones) obtained from clones was carried out by HAE-III to confirm the identity of PCR products and 12 common clones were removed from further processing to avoid sequencing of common clones (Fig. 2).
Sequence analysis and construction of phylogenetic tree
All reference sequences were obtained from the GenBank/EMBL/DDBJ/RDP [13]. Sequences (~650 bp) from the current study were analysed by the CHECK_CHIMERA program [14] to remove any chimeric rDNA clone. The similarity searches for sequences were carried out by BLAST [15] and alignment was done using CLUSTAL W [16]. Ambiguously and incorrectly aligned positions were aligned manually. Phylogenetic tree was constructed by the neighbour joining method using MEGA 4.0 software [17]. Bootstrap resampling analysis for 500 replicates was performed to estimate the confidence of tree topologies [18].
RESULTS
Sequence Similarity
A total of 60 16S rDNA clones of partial length were isolated from the rumen liquor of the goats. All the clones were subjected to sequence analysis, followed by homology search using databases: the GenBank and the Ribosomal Database Project (RDP) database. In our library, about 91.66% clones (55 clones) had ≥90% similarity to 16S rDNA data sequences from those databases. Furthermore, about 6.66% (4 clones) of the sequences were 85-89% similar to 16S rDNA data sequences, while for the remaining 1.66% (1 clone) the similarity was less than 85% (Table 1 and Table 2). Five clones (8%) displayed similarities with known bacterial species viz.
Ruminococcus albus, Ruminococcus flavefaciens, Prevotella multiformis (2 clones) and
Butyrivibrio fibrisolvens. Again five clones (8%) showed similarities with known bacterial genus viz., Ruminococcus spp, Prevotella spp and Bacillus spp (3 clones). Only a single clone had similarities with family Porphyromonadaceae (1.6%) and another clone (1.6%) exhibited similarities with phylum Firmicutes. Twenty clones (33.3%) presented similarities with Uncultured Rumen Bacteria (URB), twenty six clones (43.3%) had similarities with Uncultured Bacteria and two clones (3.3%) were similar to Uncultured Equine Intestinal Eubacteria (UEIE).
Phylogenetic Analysis
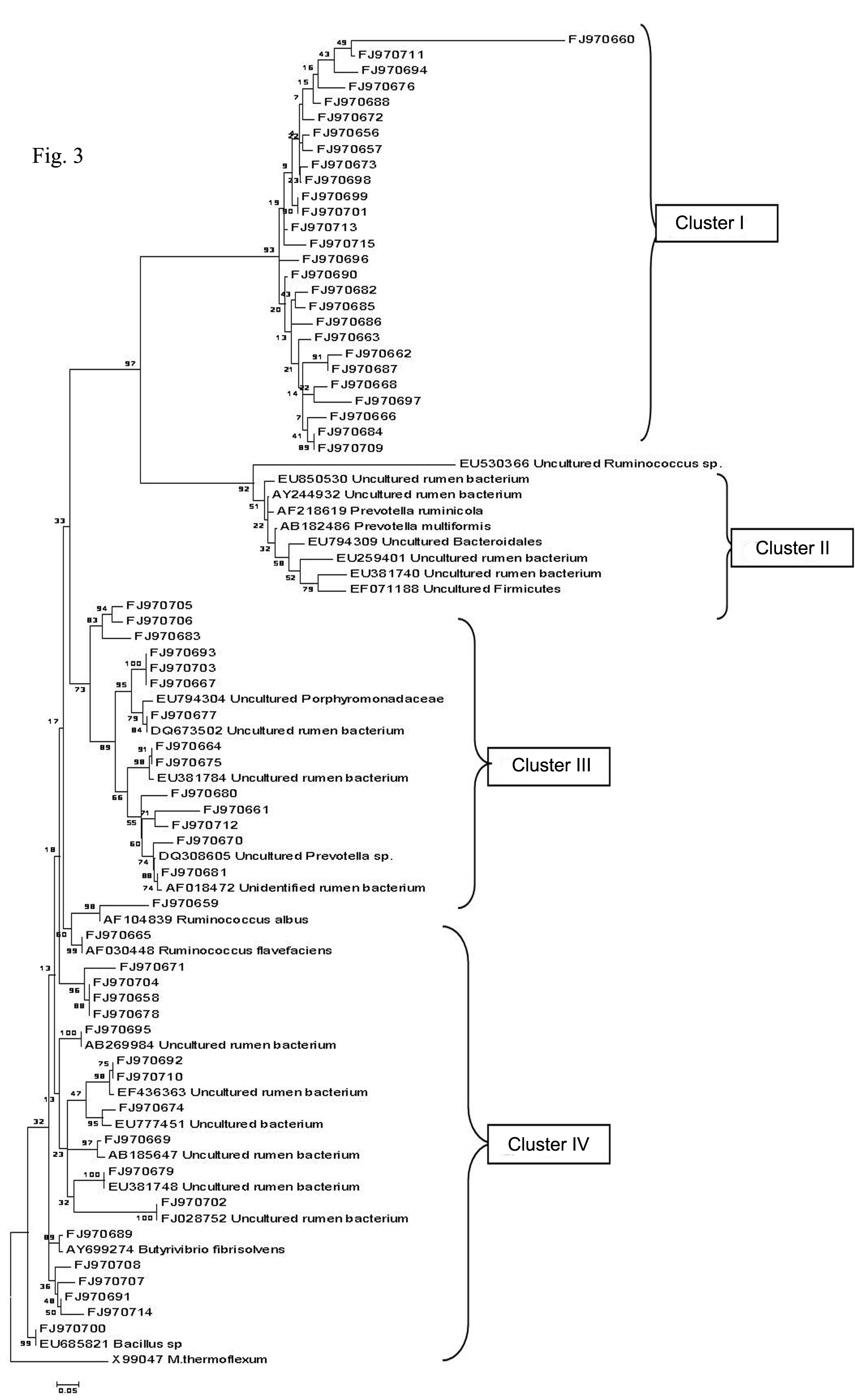
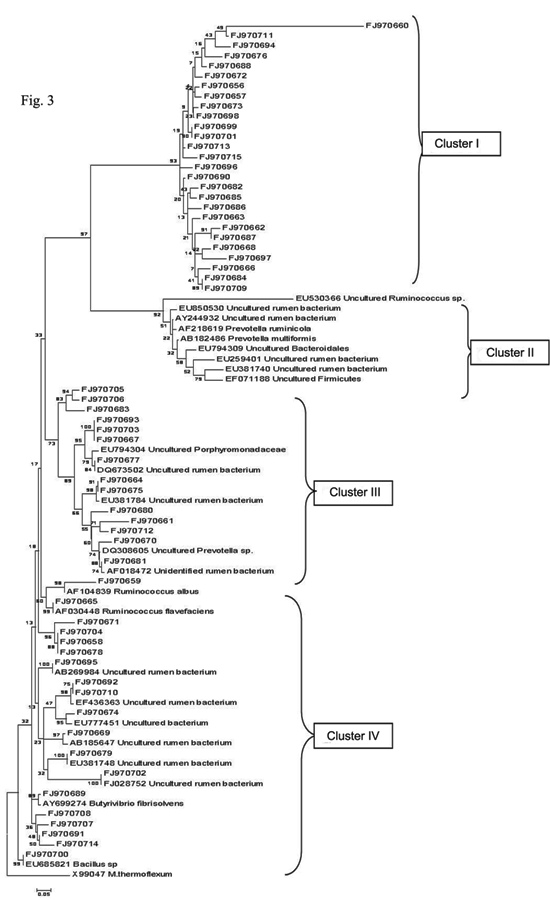
The collection of cloned 16S rRNA gene sequences of bacteria entailed several major bacterial lineages. Since the similarity for most of the sequences with those of known rumen bacteria was low to identify the sequence as representing a particular species, a phylogenetic tree was constructed to investigate their taxonomic affiliation. The phylogenetic tree was generated using MEGA 4 software to check the taxonomic identities of the 60 clones of our library. The bacterial sequences obtained from the rumen formed tightly clustered, deeply diverging groups affiliated to different bacterial phyla as well as unclassified groups. The phylogenetic tree was mainly divided in to four clusters (Fig. 3). In Cluster I, 27 clones were grouped separately and no cultured or uncultured representatives from GenBank database was grouped with these clones. Cluster II included nine reference sequences retrieved from NCBI nucleotide database which, however, did not group with any of the 60 sequences obtained in the present study. Interestingly, they were grouped adjacent to cluster I in comparison to rest of the clusters signifying lesser genetic distance between cluster I and Cluster II. Cluster III represented 14 clones which were further divided into five subgroups. Sub group-I consisted of three clones (FJ970705, FJ970706 and FJ970683) that clustered together, without showing genetic similarity with any reference sequences. Sub group-II consisted of three clones (FJ970693, FJ970703 and FJ970667) that again did not match with any of the reference sequences. Sub group-III contained one clone (FJ970677) clustered with uncultured group of family Porphyromonadaceae as well as with uncultured rumen bacterium. Sub group-IV included two clones (FJ970664 and FJ970675) clustered with uncultured rumen bacterium. Sub group-V included five clones (FJ970680, FJ970661, FJ970712, FJ970670 and FJ970681) clustered together with uncultured Prevotella as well as unidentified rumen bacterium, of which, the later two clones were at less genetic distance than the first three and thus more similar to the mentioned taxa. Cluster IV represented 19 clones that were further divided in to four subgroups. Sub group-I consisted of clones FJ970659 and FJ970665 that clustered with
Ruminococcus albus and Ruminococcus flavefaciens respectively. The same clones were also identified as
Ruminococcus albus and Ruminococcus flavefaciens by similarity search and thus confirms their identification. Sub group-II consisted of clone FJ970689 clustered with
Butyrivibrio fibrisolvens. The same clone was identified as belonging to genus Butyrivibrio by taxonomic classification at RDP which strengthened its identification as
Butyrivibrio fibrisolvens. Sub group-III consisted of clone FJ970700 clustered with genus Bacillus which was also identified up to genus (i. e. Bacillus) level by both similarity search and taxonomic classification and hence validated. Sub group-IV included remaining 15 clones, of which nine clones clustered along with one or many of themselves and six clones clustered with uncultured rumen bacteria.
Taxonomic classification using RDP
All the 60 obtained sequences were subjected to classification by using Taxonomic Classifier software available at RDP. From the highest to the lowest, the major formal taxonomic ranks obtained were domain, phylum, class, order, family, genus and species.
Classification by RDP revealed that 60 clones were mainly distributed into two phyla, namely Bacteroidetes with 21 clones (35.0 %), Firmicutes with 20 clones (33.0 %), 17 clones (29 %) fell under unclassified bacteria and two clones (3 %) were grouped under unclassified root (Table 3).
Nucleotide Sequence and Accession Numbers
The nucleotide sequence data reported in this study can be obtained from the EMBL, GenBank and DDBJ Nucleotide Sequence Databases under Accession numbers FJ970656 to FJ970715 with the generic name of AVCGRB (Anand Veterinary College Goat Rumen Bacteria).
DISCUSSION
Species identification using sequence similarity demands sequences having similarity greater than 98% [19]. In our library, 5 clones were identified as species viz.
Ruminococcus albus, Ruminococcus flavefaciens, Prevotella multiformis (2 clones) and
Butyrivibrio fibrisolvens. 21 clones belonged to Phylum Bacteroidetes and 20 clones belonged to Phylum Firmicutes. The proportions of bacterial communities in Surti goat rumen are similar to those reported previously in other rumen ecology studies. In a metagenome analysis by pyrosequencing of rumen sample of Angus-Simmental cross steers (Bos taurus) fed a diet of grass-legume hay [20], 62% of sequences were identified as the Firmicutes and 21% of sequences as the
Bacteroides fragilis group. A work published [21] reported that 52.4% of clones identified in the rumen liquor of Holstein cow (Bos taurus) fed a diet of hay belonged to the Firmicutes, and 38.1% to the Cytophaga-Flexibacter-Bacteroides (CFB) phylum. Another study [22], summarizing the published data for rumen bacteria, reported that on average 54% of rumen bacteria were members of the Firmicutes and 40% were from the CFB phylum. Yuhei et al. [23] found that 81.3% clones represented the Firmicutes, while 14.4% clones belonged to the Bacteroides fragilis group and Actinobacteria and Proteobacteria in fecal microbiota of cattle. A research group in year 2007 [24] assigned 57.1% of clones to the phylum Firmicutes, 42.2% of clones to the CFB phylum, and one clone (0.7%) to Spirochaetes in gayals (Bos gaurus frontalis, syn. B. frontalis) fed a diet composed of fresh bamboo leaves and twigs (Sinarundinaria sp.), with 50.5±3.16 % dry matter, 10.2± 0.40 % crude protein, 38.8±1.17 % crude fiber. Dowd et al. [25], using 16S rDNA bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP), evaluated ubiquitous bacteria from the cattle faeces, which included Clostridium, Bacteroides, Porpyhyromonas, Ruminococcus, Alistipes, Lachnospiraceae, Prevotella, Lachnospira, Enterococcus, Oscillospira, Cytophaga, Anaerotruncus, and Acidaminococcus spp. In a similar study [26] conducted on rumen bacterial 16S rDNA of Chinese goat, five clones (31%) showed over 97% similarities with Genbank database sequences while as eight clones (50%) showed similarities in the range of 90-96% and the remaining three clones (19%) were less than 90 % similar to unidentified bacteria. The data is comparable to the present study wherein 77% clones showed similarity in range of 95-99% with Genbank database. Further in their study, one clone (6.25%) belonged to Prevotella spp. ,while as 3 clones (5%) in our study belonged to Prevotella spp. which is again in agreement with the mentioned study. Another study in Surti buffalo [27] revealed that out of 191 clones analyzed, 41 clones (21.4%) belonged to Firmicutes ( Low G+C group), 27 clones (14.1%) belonged to CFB phylum, 13 clones (6.8%) belonged to Spirochaetes, 1 clone (0.5%) belonged to Actinobacteria and 109 clones (57%) belonged to unidentified bacteria. In present study involving goats, Actinobacteria and Spirochaetes groups were not found. This may probably indicate species difference between goat and buffalo in terms of their rumen bacterial flora. However, a study suggested [28] that sample collection time, site, method and technical bias of PCR amplification may affect the results in this kind of experiment, which possibly demands repeated attempts involving these two species to conclude the presence/absence of Actinobacteria and Spirochaetes groups of bacteria in rumen of goats.
Despite the possible deficiencies of analysis, it is concluded that surti goat rumen harbours a diverse range of bacteria. The results are more or less consistent with the previous studies. However, further studies need to be conducted to validate and extend the present observations.
ACKNOWLEDGEMENTS
Authors are thankful to the Department of Biotechnology, Government of India, for the financial assistance provided by them.
REFERENCES
-
Miron J, BenGhedalia D, Morrison
M. Invited review: adhesion mechanisms of rumen cellulolytic
bacteria. J Dairy Sci 2001;84:1294–1309.
-
Woese CR, Kandler O, Wheelis ML.
Towards a natural system of organisms: proposal for the domains
Archaea, Bacteria and Eucarya. Proc Natl Acad Sci USA
1990;87:4576–4579.
-
Hungate RE. Aroll tube method for
cultivation of strict anaerobes. In: Methods in microbiology,
Norris JR, Ribbons DW (Ed.) Vol. 3B. London and New York:
Academic Press: 1969, pp. 117–132.
-
Dehority BA, Tirabasso PA, Grifo
AP. Most-probable-number procedures for enumerating ruminal
bacteria, including the simultaneous estimation of total and
cellulolytic numbers in one medium. Appl Environ Microbiol
1989;55:2789–2792.
-
Krause DO, Russell JB. How many
ruminal bacteria are there? J Dairy Sci 1996;79:1467–1475.
-
Sylvester JT, Karnati SKR, Yu Z,
Morrison M, Firkins JL. Development of an assay to quantify
rumen ciliate protozoal biomass in cows using real-time PCR. J
Nutr 2004;134:3378–3384.
-
Shin EC, Cho KM, Lim WJ, Hong SY,
An CL, Kim EJ, Kim YK, Choi BR, An JM, Kang JM, Kim H, Yun HD.
Phylogenetic analysis of protozoa in the rumen contents of cow
based on the 18S rDNA sequences. J Appl Microbiol 2004;97:
378-383.
-
An D, Dong X, Dong Z. Prokaryote
diversity in the rumen of yak (Bos grunniens) and Jinnan cattle
(Bos taurus) estimated by 16S rDNA homology analyses. Anaerobe
2005;11:207–221.
-
Indian Livestock Census. All
India Summary Report: Livestock, Poultry, Agricultural
Machinery, Implements and Fishery Statistics, 2003.
http://dms.nic.in/ami/home.htm.
-
ICAR. Nutrient requirements of
livestock and poultry. Indian Council of Agricultural Research,
New Delhi, India, 1998.
-
Khampa S, Wanapat M,
Wachirapakorn C, Nontaso N, Wattiaux M. Effects of urea level
and sodium DL-malate in concentrate containing high cassava chip
on ruminal fermentation efficiency microbial protein synthesis
in lactating dairy cows raised under tropical conditions. Asian-
Aust J Anim Sci 2006;19:837–844.
-
Lane DJ. 16S/23S rRNA sequencing.
In: Nucleic acid techniques in bacterial systematic,
Stackebrandt E and Goodfellow M (Ed.) New York: John Wiley &
Sons: 1991, 115–175.
-
Benson DA, Karsch-Mizrachi I,
Lipman DJ, Ostelland J, Wheeler D. GenBank Nucleic Acids Res
2007;35:D1–D25.
-
Maidak BL, Cole JR, Lilburn TG,
Jr CTP, Saxman PR, Farris RJ, Garrity GM, Oslen GJ, Schmidt TM,
Tiedje JM . The RDP-II (Ribosomal Database Project). Nucl Acids
Res 2001;29: 173–174.
-
Madden TL, Tatusov RL, Zhan J.
Application of network BLAST server. Meth Enzymol
1996;266:131-141.
-
Thompson JD, Higgins DG, Gibson
TJ. CLUSTAL W: improving the sensitivity of progressive multiple
sequence alignment through sequence weighting, position-specific
gap penalties and weight matrix choice. Nucl Acids Res
1994;22:4673–4680.
-
Tamura K, Dudley J, Nei M, Kumar
S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA)
software version 4.0. Molecular Biology and Evolution
2007;24:1596–1599.
-
Felsenstein J. Confidence limits
on phylogenies: an approach using the bootstrap. Evol
1985;39:783–791.
-
Vandamme P, Pot B, Gillis M, Vos
De P, Kersters K, Swings J. Polyphasic Taxonomy, a consensus
approach to bacterial systematics. Microbiol Rev 1996;60:
407–438.
-
Brulc M, Antonopoulos D, Miller
ME, Wilsona MK, Yannarell AC, Dinsdale EA, Edwards RE, Frank ED,
Emerson JB, Wacklin P, Coutinho PM, Henrissat B, Nelson KE,
White BA. Gene-centric metagenomics of the fiber-adherent bovine
rumen microbiome reveals forage specific glycoside hydrolases.
Proc Natl Acad Sci USA 2009;6: 1948–1953.
-
Tajima K, Amnikov RI, Nagamine T,
Ogata K, Nakamura M, Matsui H, Benno Y. Rumen bacterial
diversity as determined by sequence analysis of 16S rDNA
libraries. FEMS Microbiology Ecol 1999;29:159-169.
-
Edwards JE, McEwan NR, Travis AJ,
Wallace RJ. 16S rDNA library-based analysis of ruminal bacterial
diversity. Antonie Van Leeuwenhoek 2004;86:263–281.
-
Yuhei O, Hidenori H, Mitsuo S,
Hisao I, Yoshimi B. Culture-independent analysis of fecal
microbiota in cattle. Biosci Botechnol Bochem 2005;69:1793–1797.
-
Deng W, Wanapat M, Chen S, Ma J,
Dongmei Xi, Tianbo HE, Zhifang Yang, Huaming Moa. Phylogenetic
analysis of 16S rDNA sequences manifest rumen bacterial
diversity in gayals (Bos frontalis) fed fresh bamboo leaves and
twigs (Sinarumdinaria). Asian-Aust. J Animal Sci
2007;7:1057–1066.
-
Dowd SE, Callaway TR, Wolcott RD,
Sun Y, McKeehan T, Hagevoort, Edrington TS. Evaluation of the
bacterial diversity in the feces of cattle using
16SrDNAbacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP).
BMC Microbiology 2008;8:125.
-
Yao W, Zhu W, Han Z, Antoon DL,
Barbara W, Seerp T. Analysis of rumen bacterial diversity of
goat by DGGE and 16S r DNA sequencing. Scientia Agricultura
Sinica 2004;37:1374-1378.
-
Pandya PR, Singh KM, Parnerkar S,
Tripathi AK, Mehta HH, Rank DN, Kothari RK, Joshi CG. Bacterial
diversity in the rumen of Indian Surti buffalo (Bubalus bubalis),
assessed by 16S rDNA analysis. J Appl Genet 2010, 51:395–402
-
Von Wintzingerode F, Gobel UB,
Stackebrandt E. Determination of microbial diversity in
environmental samples: pitfalls of PCR-based rRNA analysis. FEMS
Microbiol Rev 1997;21:213-22.
TABLES
Table 1. Similarity values of clones (1-30) based on 16S rDNA sequences retrieved from the rumen fluid of Indian Surti goat
|
Sr.No.
|
Clone no./ Accession no. (Query sequence)
|
Nearest relative taxa
|
Accesson no. (Subject sequence)
|
Identity (%)
|
|
1
|
FJ970656
|
Ruminococcus sp
|
EU530444
|
98
|
|
2
|
FJ970657
|
Uncultured equine intestinal eubacterium
|
AJ408183
|
96
|
|
3
|
FJ970658
|
Uncultured bacterium clone
|
FJ970704
|
92
|
|
4
|
FJ970659
|
Ruminococcus albus
|
AF104839
|
99
|
|
5
|
FJ970660
|
Uncultured bacterium clone
|
EF604814
|
99
|
|
6
|
FJ970661
|
Uncultured rumen bacterium clone
|
AB185623
|
72
|
|
7
|
FJ970662
|
Uncultured equine intestinal eubacterium
|
AJ408217
|
95
|
|
8
|
FJ970663
|
Uncultured bacterium clone
|
EU844055
|
93
|
|
9
|
FJ970664
|
Uncultured rumen bacterium clone
|
EU381784
|
99
|
|
10
|
FJ970665
|
Ruminococcus flavefaciens
|
AF030448
|
99
|
|
11
|
FJ970666
|
Uncultured bacterium clone
|
FJ172808
|
99
|
|
12
|
FJ970667
|
Uncultured rumen bacterium clone
|
FJ970703
|
97
|
|
13
|
FJ970668
|
Uncultured bacterium clone
|
FJ032376
|
99
|
|
14
|
FJ970669
|
Uncultured bacterium clone
|
AB185647
|
99
|
|
15
|
FJ970670
|
Uncultured bacterium clone
|
EU473041
|
99
|
|
16
|
FJ970671
|
Uncultured bacterium clone
|
FJ172887
|
93
|
|
17
|
FJ970672
|
Uncultured bacterium clone
|
EU845633
|
88
|
|
18
|
FJ970673
|
Uncultured bacterium clone
|
EU381740
|
92
|
|
19
|
FJ970674
|
Uncultured bacterium clone
|
EU777451
|
85
|
|
20
|
FJ970675
|
Uncultured rumen bacterium clone
|
EU381784
|
99
|
|
21
|
FJ970676
|
Uncultured Firmicutes
|
EF071188
|
97
|
|
22
|
FJ970677
|
Uncultured rumen bacterium clone
|
FJ970704
|
99
|
|
23
|
FJ970678
|
Uncultured rumen bacterium clone
|
FJ970658
|
99
|
|
24
|
FJ970679
|
Uncultured bacterium clone
|
EU843043
|
94
|
|
25
|
FJ970680
|
Prevotella sp.
|
DQ308605
|
99
|
|
26
|
FJ970681
|
Unidentified rumen bacterium
|
AF018472
|
97
|
|
27
|
FJ970682
|
Uncultured bacterium clone
|
EU850530
|
95
|
|
28
|
FJ970683
|
Uncultured bacterium clone
|
EU845295
|
99
|
|
29
|
FJ970684
|
Prevotella multiformis
|
AB182486
|
99
|
|
30
|
FJ970685
|
Uncultured rumen bacterium clone
|
FJ970685
|
95
|
Table 2. Similarity values of clones (31-60) based on 16S rDNA sequences retrieved from the rumen fluid of Indian Surti goat
|
Sr.No.
|
Clone no./ Accession no. (Query sequence)
|
Nearest valid relative
|
Accesson no. (Subject sequence)
|
Identity (%)
|
|
31
|
FJ970686
|
Uncultured rumen bacterium clone
|
EU381955
|
91
|
|
32
|
FJ970687
|
Uncultured rumen bacterium clone
|
EU259401
|
98
|
|
33
|
FJ970688
|
Uncultured rumen bacterium clone
|
EU843421
|
95
|
|
34
|
FJ970689
|
Uncultured rumen bacterium clone
|
EU259487
|
99
|
|
35
|
FJ970690
|
Uncultured rumen bacterium clone
|
AY244932
|
99
|
|
36
|
FJ970691
|
Uncultured bacterium clone
|
EU844715
|
98
|
|
37
|
FJ970692
|
Uncultured rumen bacterium clone
|
EU842956
|
99
|
|
38
|
FJ970693
|
Uncultured Porphyromonadaceae
|
EU794304
|
91
|
|
39
|
FJ970694
|
Uncultured bacterium clone
|
EU259402
|
97
|
|
40
|
FJ970695
|
Uncultured rumen bacterium clone
|
AB269984
|
99
|
|
41
|
FJ970696
|
Uncultured rumen bacterium clone
|
EF4363911
|
88
|
|
42
|
FJ970697
|
Uncultured bacterium clone
|
EU843262
|
98
|
|
43
|
FJ970698
|
Uncultured bacterium clone
|
EU843171
|
97
|
|
44
|
FJ970699
|
Bacillus sp.
|
AY461750
|
99
|
|
45
|
FJ970700
|
Bacillus sp.
|
EU685821
|
99
|
|
46
|
FJ970701
|
Bacillus sp.
|
GQ120667
|
99
|
|
47
|
FJ970702
|
Uncultured rumen bacterium clone
|
FJ028752
|
97
|
|
48
|
FJ970703
|
Uncultured rumen bacterium clone
|
FJ970667
|
99
|
|
49
|
FJ970704
|
Uncultured rumen bacterium clone
|
FJ970658
|
99
|
|
50
|
FJ970705
|
Uncultured bacterium clone
|
FJ172882
|
97
|
|
51
|
FJ970706
|
Uncultured bacterium clone
|
EU459539
|
98
|
|
52
|
FJ970707
|
Uncultured rumen bacterium clone
|
AB185576
|
94
|
|
53
|
FJ970708
|
Uncultured bacterium clone
|
DQ810117
|
94
|
|
54
|
FJ970709
|
Prevotella multiformis
|
AB182486
|
99
|
|
55
|
FJ970710
|
Uncultured bacterium clone
|
FJ970692
|
97
|
|
56
|
FJ970711
|
Butyrivibrio fibrisolvens
|
U77341
|
99
|
|
57
|
FJ970712
|
Uncultured bacterium clone
|
AY578359
|
99
|
|
58
|
FJ970713
|
Uncultured bacterium clone
|
AY244958
|
99
|
|
59
|
FJ970714
|
Uncultured bacterium clone
|
EU843442
|
87
|
|
60
|
FJ970715
|
Uncultured bacterium clone
|
EU842405
|
99
|
Table 3. Summary of sequence similarity at 95% confidence level by Taxonomic Classifier (Ribosomal Database Project)
|
S.No.
|
Description
|
Percentage
|
GenBank Accession numbers
|
|
Clones with species level identification
|
|
Nil
|
|
Clones with genus level identification
|
|
I
|
Genus Prevotella (2)
|
3.0
|
FJ970684, FJ970709
|
|
II
|
Genus Butyrivibrio (2)
|
3.0
|
FJ970689, FJ970711
|
|
III
|
Genus Ruminococcus (1)
|
1.6
|
FJ970665
|
|
IV
|
Genus Succiniclasticum (1)
|
1.6
|
FJ970688
|
|
V
|
Genus Bacillus (3)
|
5.0
|
FJ970699, FJ970700, FJ970701
|
|
Clones with family level
identification
|
|
I
|
Unclassified Prevotellaceae (6)
|
10.0
|
FJ970668, FJ970680,
FJ970682, FJ970685, FJ970690,
FJ970712
|
|
II
|
Unclassified Lachnospiraceae (6)
|
10.0
|
FJ970656, FJ970673,
FJ970691, FJ970696, FJ970698,
FJ970708
|
|
III
|
Unclassified Ruminococcaceae (4)
|
6.0
|
FJ970660, FJ970676, FJ970679,
FJ970715
|
|
Clones with order level
identification
|
|
I
|
Unclassified Bacteroidales (10)
|
16.0
|
FJ970663, FJ970664, FJ970666, FJ970667,
FJ970675, FJ970677, FJ970681, FJ970693, FJ970703, FJ970705
|
|
II
|
Unclassified Clostridiales (2)
|
3.0
|
FJ970657, FJ970707
|
|
Clones with class level
identification
|
|
I
|
Unclassified Bacteroidetes (3)
|
5.0
|
FJ970687, FJ970705, FJ970706
|
|
II
|
Unclassified Clostridia (1)
|
1.6
|
FJ970713
|
|
Clones with phylum level
identification
|
|
I
|
Unclassified Bacteria (17)
|
28.0
|
FJ970658, FJ970662, FJ970669, FJ970671,
FJ970672, FJ970674, FJ970678, FJ970683, FJ970686, FJ970692,
FJ970694, FJ970695, FJ970697, FJ970702, FJ970704, FJ970710,
FJ970714
|
|
Clones with root level
identification
|
|
I
|
Unclassified root (2)
|
3.0
|
FJ970659, FJ970661
|
FIGURES
Fig. 1: Agarose gel electrophoresis of M13 PCR products showing recombinant as well as non recombinant clones. +ve clones represent the recombinant clones while as –ve clones represent the non-recombinant clones.
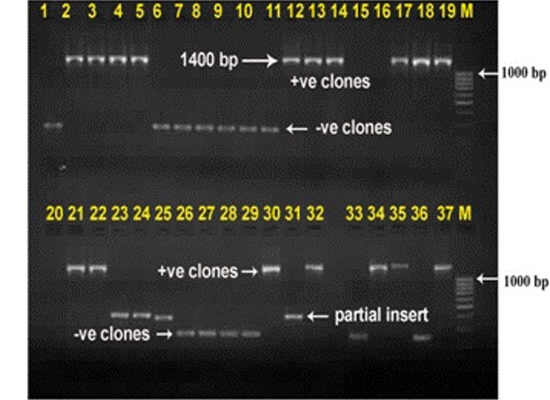
Fig. 2: RFLP by HAE-III digestion. Twelve clones with common banding pattern (clones no. 4, 11, 38, 23, 45, 31, 65, 32, 39, 61, 50 and 63) were removed to screen out the common clones.
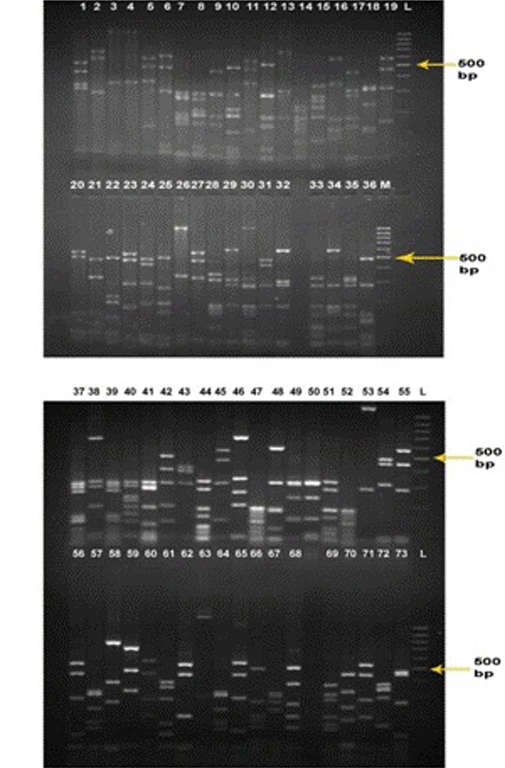
Fig. 3: Phylogenetic relationships of partial 16S rDNA sequences of clones recovered from Surti goat rumen fluid samples. The unrooted tree was inferred by the neighbour joining method using the MEGA 4 software. The scale bar equals to an average of 5 nucleotide substitutions per 100 positions. The 16S rDNA sequences of the present study are represented by ‘FJ’. Cluster I involves 27 novel clones that grouped seperately, Cluster II involves 9 reference 16S rDNA sequences from Genbank that did not group with any of the 60 clones, Cluster III and IV involved 14 and 19 clones respectively that grouped or did not group with any of the representative sequences from genbank.
|
 |